
Why Pharmacovigilance Regulations feel dry and confusing?
A Mental Model to understand Pharmacovigilance Regulations without memorizing numbers

I believe everyone will agree that terms like GVP Module IV, 21 CFR 314.80 sound dry and confusing. They feel authoritative but also abstract.
For someone new to PV, the natural tendency is to start memorizing names before understanding intent. People begin following documents, but seldom reason about why those documents exist in the first place.
The funny thing is that with experience, the confusion doesn’t disappear. Experienced professionals often know which document to refer to and understand what is expected operationally. Yet, many still feel uneasy during audits or inspections. This uneasiness usually comes from the fact that regulations are remembered as documents, not as principles.
Pharmacovigilance regulations are frequently described as complex. In reality, the same safety activity is often described differently in the EU and the US. It appears in different documents and is referenced using different numbers. This creates the impression that the rules themselves are changing, when in fact only the structure is.
In my experience, this fragmentation is one of the biggest sources of confusion in Pharmacovigilance.
Faced with this fragmentation, most professionals develop a survival mechanism:
Memorize module numbers
Bookmark CFR sections
Keep guidance documents handy
But when faced with deeper questions like why a process exists or which rule applies in a new situation or how two regulations relate, there is fumble and uncertainity.
What are regulations really trying to do?
The fact is that regulations were not written to make our lives difficult, nor to create documents for auditors to check. They were written because something went wrong, often repeatedly, and society decided that “good intentions” were not enough.
At their core, pharmacovigilance regulations are about controlling uncertainty. Reports, timelines, or formats are the means to achieve this control.
When a medicine is released into the real world, uncertainty explodes:
Patients are different from clinical trial participants
Doses are missed, changed, or misunderstood
Drugs are combined in ways never studied
Rare events finally start appearing
Risks can not be avoided but the effort should be to detect them early and acted upon responsibly.
That is the real purpose of pharmacovigilance regulations.
Every regulation, no matter how complex, is trying to answer a few fundamental questions:
How will you find safety problems?
How fast will you react when you find them?
How will you decide what matters and what doesn’t?
How will you prove that you did the right thing at the right time?
This is why regulations inevitably give rise to systems, responsibilities, documentation, timelines, and oversight.
Regulations are the guardrails for decision-making under uncertainty
Laws vs Regulations vs Guidance
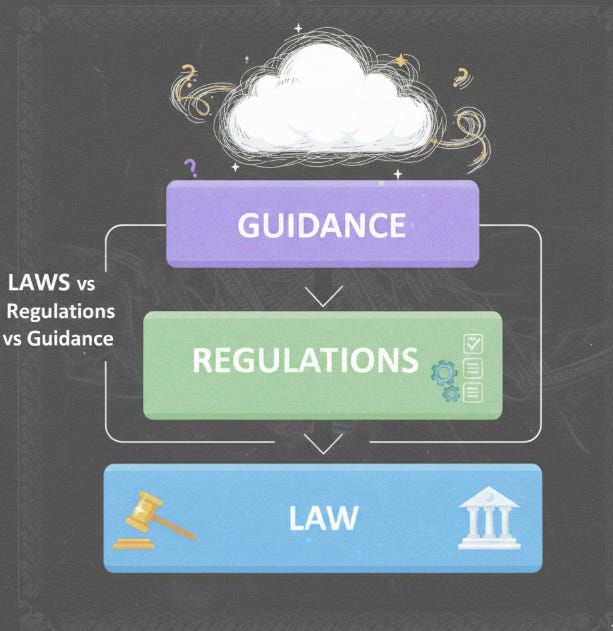
One of the reasons Pharmacovigilance regulations feel confusing is terminology. Words like law, regulation, and guidance are often used interchangeably but in reality, they are very different, and each carries a different weight in practice.
Law
A law is passed by a government body. It is binding on everyone within its jurisdiction. Laws are the “ground rules”. If you break them, there will be legal consequences.
Regulation
A regulation is a specific requirement derived from a law. It provides the detailed instructions for how the law should be implemented. In PV, this is where you’ll find concrete requirements on reporting timelines, case definitions, and responsibilities.
Guidance
Guidance documents are written by regulators to explain how they expect the law or regulation to be interpreted or applied. They are not legally binding, but failing to follow them can raise questions during inspections or audits.
This distinction explains why the same safety activity may be described in multiple documents, using slightly different language, and why some requirements “feel optional” while others are enforceable.
Two Regulatory Philosophies: EU vs US

As discussed, regulations have a single goal: Protecting patients by managing the uncertainty inherent in medicine use.
The way this goal is structured on paper can look very different depending on where you are. Some regions focus on flexible frameworks and modular guidance, while others emphasize strict legal obligations and prescriptive rules.
At a high level, two regulatory philosophies dominate the world of PV:
The EU Approach – Modular, lifecycle-oriented
The US Approach – Legal, obligation-driven
The EU Approach
In Europe, regulators separate what must happen from how it should happen operationally. The law sets the obligations while operational expectations are explained in GVP modules and guidance documents. GVP refers to Good Pharmacovigilance Practises. Each module focuses on a different part of the PV lifecycle: from organization, responsibilities, and systems, to signal detection and risk management.
The US Approach
In the United States, regulators tend to embed operational detail directly into the law itself. Legal texts specify obligations, timelines, and responsibilities. Guidance documents exist, but they primarily clarify rather than introduce new expectations. Compliance is measured directly against the law and deviations are scrutinized.
Now lets jump into details for each of these approaches:
The EU Approach: Law + GVP Modules
In the European system, pharmacovigilance is structured as a set of layers.
At the top sits the law. This law establishes the obligation: if you market a medicine, you are responsible for monitoring its safety throughout its lifecycle. It does not tell you exactly how to do this day to day. Instead, it defines what must be ensured.
Then comes the GVP modules to translate legal intent into practical expectations. Think of them as functional building blocks of a pharmacovigilance system.
Each module answers a specific operational question:
How should a company organize itself for pharmacovigilance?
How should safety data be collected and managed?
How are signals identified, evaluated, and acted upon?
How are risks minimized and communicated?
How is compliance demonstrated and inspected?
Instead of embedding all these details directly into law, the EU chose a modular approach. This allows the system to evolve. Modules can be updated, expanded, or clarified as science, technology, and real-world experience change, without rewriting the law every time.
Currently EMA has defined GVP Modules as below:
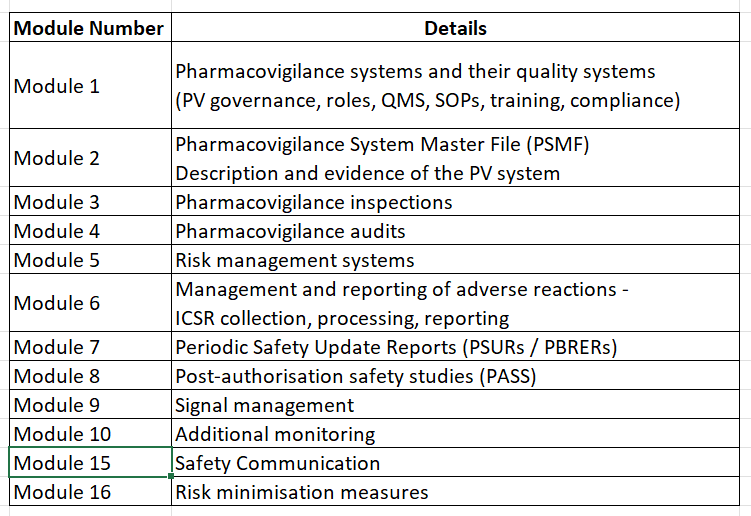
Note that as of today, the Module 11,12,13 and 14 are void. The details can be found on official EMA site.
Any pharmacovigilance activity in an organization will be a part of one of these modules.
The US Approach: CFR + FDA Guidance
In the United States, pharmacovigilance is not primarily explained through modules or operational guidance layered on top of the law. Instead, the law itself carries much of the operational weight.
US pharmacovigilance requirements are written into the Code of Federal Regulations (CFR). These are legally binding rules which define:
What must be reported
By when it must be reported
In what format
By whom
The expectation is that if something is written in the CFR, it is non-negotiable. Guidance documents do exist in the US system, but their role is narrower than in the EU. Their role is to clarify the regulation by providing examples or best practises.
Some of the sections of CFR are summarized below:
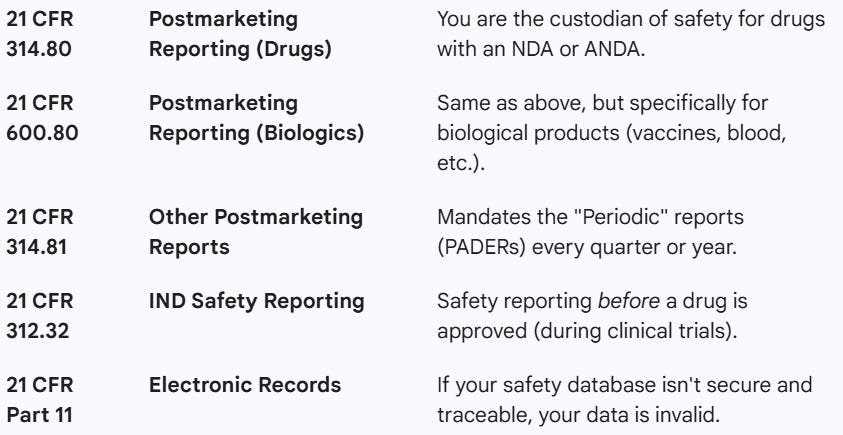
The 3 important guidance documents from FDA are as follows:
Postmarketing Adverse Event Reporting for Human Drug and Biological Products
Good Pharmacovigilance Practices and Pharmacoepidemiologic Assessment
Providing Submissions in Electronic Format - Postmarketing Safety Reports
A Unifying mental model: What all PV regulations are actually asking
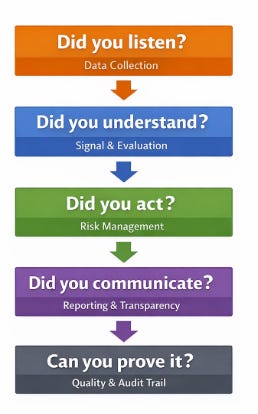
Despite structural differences, the US and EU systems are asking the same questions:
Did you listen? (Data Collection)
Are you systematically collecting safety information from all relevant sources?Did you understand? (Signal & Evaluation)
Did you evaluate the information properly and recognize what matters?Did you act? (Risk Management)
When a risk was identified, did you take timely and appropriate action?Did you communicate? (Reporting & Transparency)
Did the right people receive the right information at the right time?Can you prove it? (Quality & Audit Trail)
Is there objective evidence that these steps happened as claimed?
If you can confidently answer these five questions, you are already aligned with the intent of most pharmacovigilance regulations, regardless of how they are written.
Regulations feel fragmented because each question above is often addressed in different documents.
A powerful mental model to understand regulations is to stop focusing on questions like “Which module covers this?” or “Which CFR section do I quote?”. Instead, try to answer “Which of the five questions am I answering right now?”
You will realize that:
Case processing answers Did you listen?
Signal detection answers Did you understand?
Risk management answers Did you act?
Expedited reporting answers Did you communicate?
PSMF, audits, and inspection readiness answer Can you prove it?
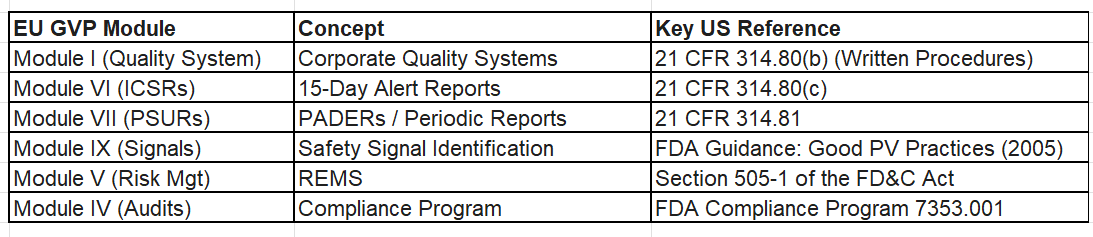
Below is a mapping between EU’s GPPV moduels and CFR’s sections:
Case Study
Step 1: A patient experiences an adverse event(AE)
A patient taking a marketed medicine experiences an unexpected side effect. It may or may not be serious. At this point, no conclusions are needed. Only one thing matters: Is there a mechanism to hear about this event? This immediately maps to the first fundamental regulatory question: Did you listen?
Step 2: The Patient Contacts a Call Center
The patient calls a company-operated call center to report what happened. The regulation at this point would be interested in if the information was captured accurately and if it was recognized as a potential safety report. At this point, the organization is still answering: Did you listen?
It is a data entry point into the PV system.
EU GVP: Module VI – Management and Reporting of ICSRs
(All sources must be capable of capturing safety information)US CFR:21 CFR 314.80(b) – Reporting requirements
(Applies regardless of source: call centers, sales reps, emails, etc.)
Step 3: Recognition of a Safety Case
The call center agent identifies that the information meets the minimum criteria to be considered a valid adverse event report. The system must now transfer the data to the pharmacovigilance function.
EU GVP: Module VI – Case validation and handling
US CFR: 21 CFR 314.80(a) and (b) – Definition and scope of reportable events
Step 4: Case Processing and Documentation
The report is entered into the safety database. Details are clarified where possible. Dates, seriousness, outcomes, and product information are recorded. At this stage, the organization is preparing to answer two regulatory questions:
Did you listen? (completeness and traceability)
Can you prove it? (audit trail and records)
EU GVP: Module VI – Case processing
Module I – PV quality system and documentation expectationsUS CFR: 21 CFR 314.80(e) – Records and reports
(Retention, traceability, and retrievability)
Step 5: Medical Evaluation and Initial Assessment
Now the case is assessed by qualified personnel for seriousness, expectedness, and causality. Medical resoning is expected at this stage. It answers the question “Did you understand?”
EU GVP:
Module VI – Medical review
Module I – Defined roles and responsibilitiesUS CFR:
21 CFR 314.80(c) – Evaluation of postmarketing reports
(Medical judgment applied to reporting decisions)
Step 6: Determination of Reporting Timelines
Based on assessment, the organization determines whether expedited reporting is required and initiates submission within required timelines. This is where regional differences become visible on paper. At this step, the question being answered is: Did you communicate?
EU GVP: Module VI – Expedited reporting timelines
US CFR: 21 CFR 314.80(c)(1) – 15-day Alert reports
(and follow-up obligations)
Step 7: Aggregation and Signal Detection
One case rarely changes the benefit–risk profile of a medicine. Over time, patterns are examined such as Are similar events appearing?Is the frequency changing?Is the severity evolving? The case is not viewed in isolation. It becomes part of cumulative safety data reviewed over time. The organization is trying to answer: Did you understand (beyond the individual case)?
EU GVP:
Module IX – Signal Management
Module VII – Periodic Safety Update ReportsUS CFR:
21 CFR 314.80(c)(2) – Periodic reporting
(Aggregate review supported by FDA guidance)
Step 8: Decision-Making and Risk Management
Suppose analysis suggests a new or changing risk. Decisions are made: labeling updates, risk minimization, or further studies. Regulations are not concerned about dictating what decision to be made. They are concerned that a decision must be made, justified, and documented. The question which is being answered at this step is: Did you act responsibly?
EU GVP:
Module V – Risk Management Systems
Module VIII – PASS (if needed)US CFR:
Risk mitigation expectations embedded in law, supported by guidance
(e.g., labeling changes, postmarketing requirements)
Step 9: Safety Communication
If action is required, information must be communicated to regulators, healthcare professionals, or patients.
EU GVP:
Module XI – Public Communication
Module XV – Safety CommunicationUS CFR:
Communication obligations embedded in reporting and labeling frameworks
This step ensures the organization can answer:
Did the right people know, at the right time, in the right way?
Step 10: Inspection, Audit, and Proof
Later, an inspector reviews the case. At this point, the question is no longer what happened, but how you handled it.
EU GVP:
Module I – PV System & Quality
Module II – PSMF
Module III – Inspections
Module IV – AuditsUS CFR:
21 CFR 314.80(j) – FDA inspection authority
(plus broader inspection provisions)
This single adverse event, touched multiple EU GVP modules and multiple US CFR obligations. Now, instead of focuing on “documents”, start focusing on “intent”.
Ask yourself:
Am I listening for safety information?
Am I trying to understand what this information means?
Am I deciding or acting based on that understanding?
Am I communicating something externally?
Am I creating evidence that this happened correctly?
Once you identify which question you are answering, the relevant regulation, EU module or US CFR section, almost always becomes obvious. Using this mental model, the understanding of regulations becomes simple.